
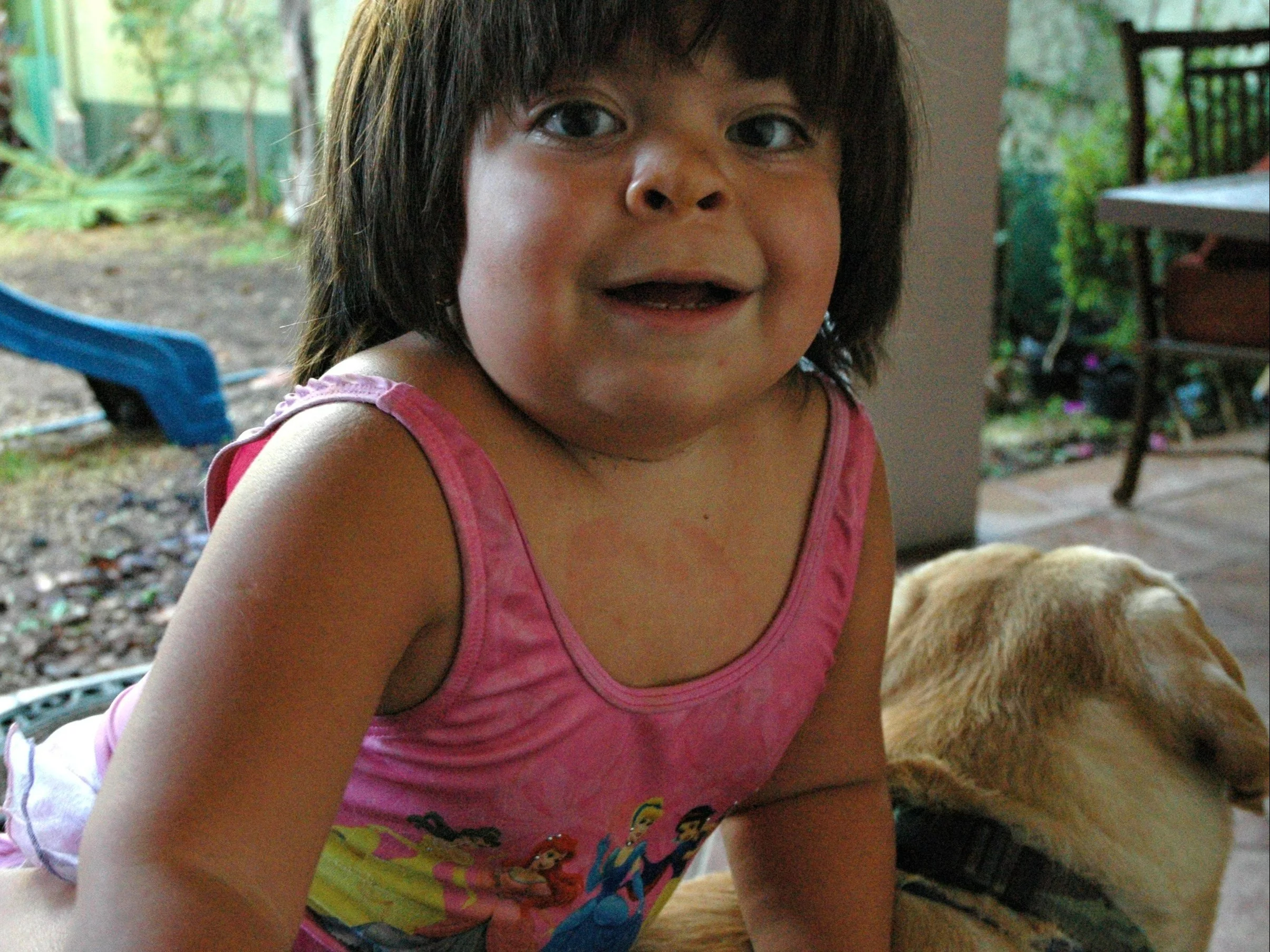
Mukopolisacharydoza (w skrócie MPS) to rzadka choroba genetyczna, która powoduje nieprawidłowości w przebiegu procesów przemiany materii. Do rozwoju choroby dochodzi na skutek błędu genetycznego, który powoduje, że w organizmie gromadzą się mukopolisacharydy. Związki te wchodzą w skład tkanki łącznej. Ich budowa uwzględnia łańcuchy cząsteczek cukrów. U osób zdrowych mukopolisacharydy są rozkładane w lizosomach przez odpowiednie enzymy. W przypadku mukopolisacharydozy dochodzi do zaburzeń w procesie metabolizowania mukopolisacharydów, które mają związek z brakiem lub nieprawidłowościami w budowie rozkładających je enzymów. Na skutek zaburzeń o podłożu genetycznym produkty rozpadu mukopolisacharydów gromadzą się w lizosomach, powodując uszkodzenia komórek.
Mukopolisacharydoza i inne choroby rzadkie powodują poważne trudności diagnostyczne. Warto dowiedzieć się więcej na temat przyczyn, objawów i leczenia mukopolisacharydozy, której pierwsze objawy często są niewłaściwie diagnozowane, co prowadzi do pogorszenia się stanu zdrowia chorych, powodując m.in. niesprawność ruchową.
Jak często występuje mukopolisacharydoza?
Mukopolisacharydoza występuje u 1 na 100 000 żywo urodzonych dzieci. Zaliczana jest do grupy rzadkich chorób metabolicznych o podłożu genetycznym. Występuje pod wieloma postaciami, prowadząc do stopniowego wyniszczenia organizmu chorego.
U osób zdrowych mukopolisacharydy metabolizowane są w lizosomach dzięki ponad dwudziestu różnym enzymom. Jeżeli, wskutek błędu genetycznego, brakuje w organizmie któregoś z enzymów lub działa on w wadliwy sposób, to dochodzi do zaburzeń procesów metabolicznych i rozwoju mukopolisacharydozy.
Mukopolisacharydoza diagnozowana jest w kilku typach. Przebieg choroby i spektrum jej objawów ma związek z brakiem lub nieprawidłowym działaniem konkretnego enzymu. Nie mamy więc do czynienia z jedną chorobą – wyróżnia się wiele mukopolisacharydoz, a każda z nich wiąże się z nieco innymi rokowaniami.
Typy mukopolisacharydozy
Mukopolisacharydoza to choroba wieloukładowa, której objawy dotyczą różnych narządów. Ze względu na dużą różnorodność i brak powiązania pomiędzy poszczególnymi objawami, pacjenci często przez długie lata czekają na prawidłową diagnozę, co nie pozostaje bez wpływu na ich stan zdrowia. W wielu przypadkach MPS diagnozowana jest jako niskorosłość.
Dotychczas zidentyfikowano 11 różnych niedoborów enzymatycznych, które powodują siedem odrębnych typów mukopolisacharydozy. W wyniku niedoboru poszczególnych enzymów w lizosomach różnych komórek ludzkiego organizmu gromadzą się uszkadzające je glikozaminoglikany.
Klasyfikacja mukopolisacharydoz uwzględnia:
- MPS I H (choroba Hurlera), MPS I S (choroba Scheiego), MPS I H-S (choroba Hurler–Scheiego), MPS II A (choroba Huntera – postać ciężka), MPS II B (choroba Huntera – postać łagodna) – typy mukopolisacharydozy powodujące nadmierne gromadzenie się w lizosomach siarczanu heparanu i siarczanu dermatanu,
- MPS III A (choroba Sanfilippo typ A), MPS III B (zespół Sanfilippo typ B), MPS III C (zespół Sanfilippo typ C), MPS III D (zespół Sanfilippo typ D) – typy mukopolisacharydozy powodujące, że w lizosomach gromadzi się siarczan heparanu,
- MPS IV A (choroba Morquio typ A) – wywołana przez brak lub niedobór enzymu o nazwie sulfataza 6–galaktozowa – ten typ mukopolisacharydozy powoduje, że w lizosomach gromadzi się siarczan keratanu oraz chondroityno–6–siarczan,
- MPS IV B – (choroba Morquio typ B) – ten typ mukopolisacharydozy powoduje, że w lizosomach gromadzi się siarczan keratanu,
- MPS VI (choroba Maroteaux-Lamy’ego) – ten typ mukopolisacharydozy powoduje, że w lizosomach gromadzi się siarczan dermatanu,
- MPS VII (choroba Sly’a) – ten typ mukopolisacharydozy powoduje, że w lizosomach gromadzi się siarczan heparanu, siarczan dermatanu i chondroityno–6–siarczan,
- MPS IX (choroba Natowicza) – ten typ mukopolisacharydozy powoduje, że w lizosomach gromadzi się kwas hialuronowy.
W jaki sposób dziedziczona jest mukopolisacharydoza?
Większość mukopolisacharydoz dziedziczona jest w sposób autosomalny recesywny. Dziedziczenie mukopolisacharydozy typu II jest sprzężone z chromosomem X.
Jak wykryć mukopolisacharydozę?
Mukopolisacharydoza to choroba powodująca trudności diagnostyczne. Wstępną metodą jest ilościowe oznaczenie mukopolisacharydów w moczu oraz określenie ich rodzaju. Warto wiedzieć, że ilość mukopolisacharydów w moczu zarówno u osób chorych, jak i zdrowych zależy od wielu czynników np. wieku i stężenia moczu, dlatego niekiedy badanie należy powtórzyć, gdy np. wynik znajduje się w granicach normy. W celu m.in. określenia nosicielstwa stosowana jest identyfikacja mutacji, czyli analiza DNA izolowanego np. z krwi pacjenta.
Możliwe jest wykrycie mukopolisacharydozy jeszcze na etapie życia płodowego – na etapie badań prenatalnych wykonywana jest analiza płynu owodniowego.
Objawy mukopolisacharydozy
Objawy, które mogą wskazywać na mukopolisacharydozę, to m.in.:
- diagnozowana w okresie niemowlęcym przepuklina pępkowa i pachwinowa,
- polipy błony śluzowej i częste infekcje górnych dróg oddechowych,
- zmętnienie rogówki,
- osłabienie słuchu,
- nadmierna pobudliwość i ruchliwość dzieci,
- zaburzenia ze strony centralnego układu nerwowego,
- zmiany kostne powodujące deformacje ciała m.in. w obrębie klatki piersiowej.
Inne objawy kliniczne mukopolisacharydozy to m.in.:
- zaburzenia wzrostu (niedobór wzrostu),
- choroby serca,
- sztywność i ból stawów – znaczące zajęcie układu kostnego prowadzi do niepełnosprawności w późniejszym życiu,
- zmiany w obrębie tkanki podskórnej,
- niestabilność kręgosłupa szyjnego,
- wady zgryzu,
- w niektórych typach MPS upośledzenie umysłowe.
Jak wygląda leczenie mukopolisacharydozy?
Brak jest możliwości przyczynowego leczenia mukopolisacharydozy. Nie jest również możliwe całkowite wyleczenie tej choroby. Testowane metody leczenia z wykorzystaniem m.in. transfuzji krwi, które miały uzupełnić niedobory enzymów brakujących w organizmie chorego, nie przyniosły oczekiwanych rezultatów. U niektórych pacjentów (mukopolisacharydoza typ II) poprawę stanu zdrowia zapewnia przeszczep szpiku kostnego, jednak jest to terapia ryzykowna.
Nowoczesną metodą leczenia mukopolisacharydozy jest enzymatyczna terapia zastępcza, która polega na bezpośrednim dożylnym podawaniu oczyszczonego enzymu. W ten sposób leczona jest np. mukopolisacharydoza typu I, II i VI. Zastępcza terapia enzymatyczna nie jest jednak sposobem na wyleczenie mukopolisacharydozy – jedynie łagodzi objawy i przebieg choroby.
Najczęściej stosowane jest leczenie objawowe, które dopasowane jest do indywidualnych potrzeb każdego pacjenta, uwzględniając m.in. zabiegi chirurgiczne, podawanie leków uspokajających, przeszczep rogówki, podawanie leków, które osłabią ruch jelit, podawanie leków znoszących objawy neurologiczne np. napady drgawkowe, stosowanie odpowiedniej diety. Istotną rolę w leczeniu pacjentów z mukopolisacharydozą odgrywa rehabilitacja.
Czytaj też:
Choroby rzadkie: postawienie diagnozy, znalezienie leczenia to sukces i satysfakcjaCzytaj też:
Leków coraz więcej, ale Polska z opóźnieniami. Problem? Wysokie koszty
Źródła:
- chorobyrzadkie.pl
- A. Antczak, M. Myśliwiec, P. Pruszczyk, Wielka Interna, Wydanie II Tom 9, Reumatologia Puszczewicz Mariusz, Medical Tribune Polska, Warszawa, 2016
- K. Prusek, Eugeniusz J. Kucharz, Enzymatyczna terapia zastępcza w leczeniu mukopolisacharydoz, Reumatologia 2011, 49, 2, s. 122–125